Lamin research
Introduction
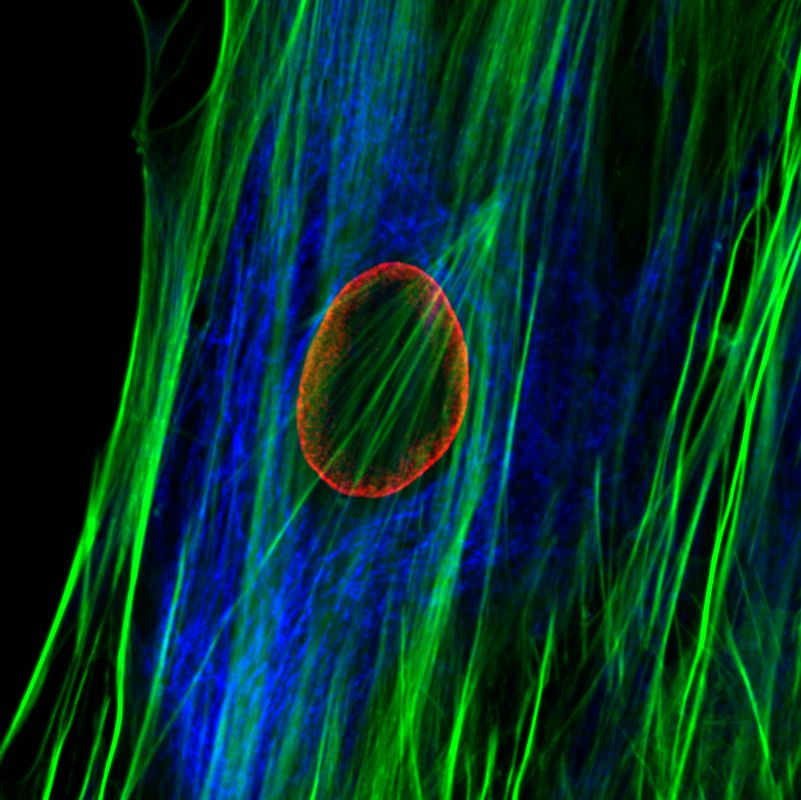
The nuclear lamins are type V intermediate filament (IF) proteins. In the cell nucleus, the lamins form the nuclear lamina, a protein meshwork between the peripheral heterochromatin and the inner nuclear membrane. The lamins are involved in multiple nuclear functions. They play a role in determining the nuclear size and shape, and influence chromatin organization and gene activity by binding to chromatin. Lamins have also been implicated in DNA replication, transcription and epigenetic regulation of chromatin. More than 400 different mutations in the LMNA gene encoding A-type lamins (primarily lamin A and C) cause a group of disease known as “laminopathies”. These include cardiomyopathies, muscular dystrophies, lipodystrophy, axonal neuropathy and premature ageing disease Hutchinson-Gilford progeria syndrome (HGPS).
Dilated cardiomyopathy (DCM) is a progressive myocardial disease characterized by reduced systolic function and dilatation of left or both cardiac ventricles. DCM causes considerable morbidity and mortality, and it is the leading cause of heart transplantation. Approximately 30-50% of the DCM cases are genetic. DCM-related mutations have been reported in up to 50 genes and the second most common gene affected is LMNA (Rosenbaum et al., 2020).
Our research
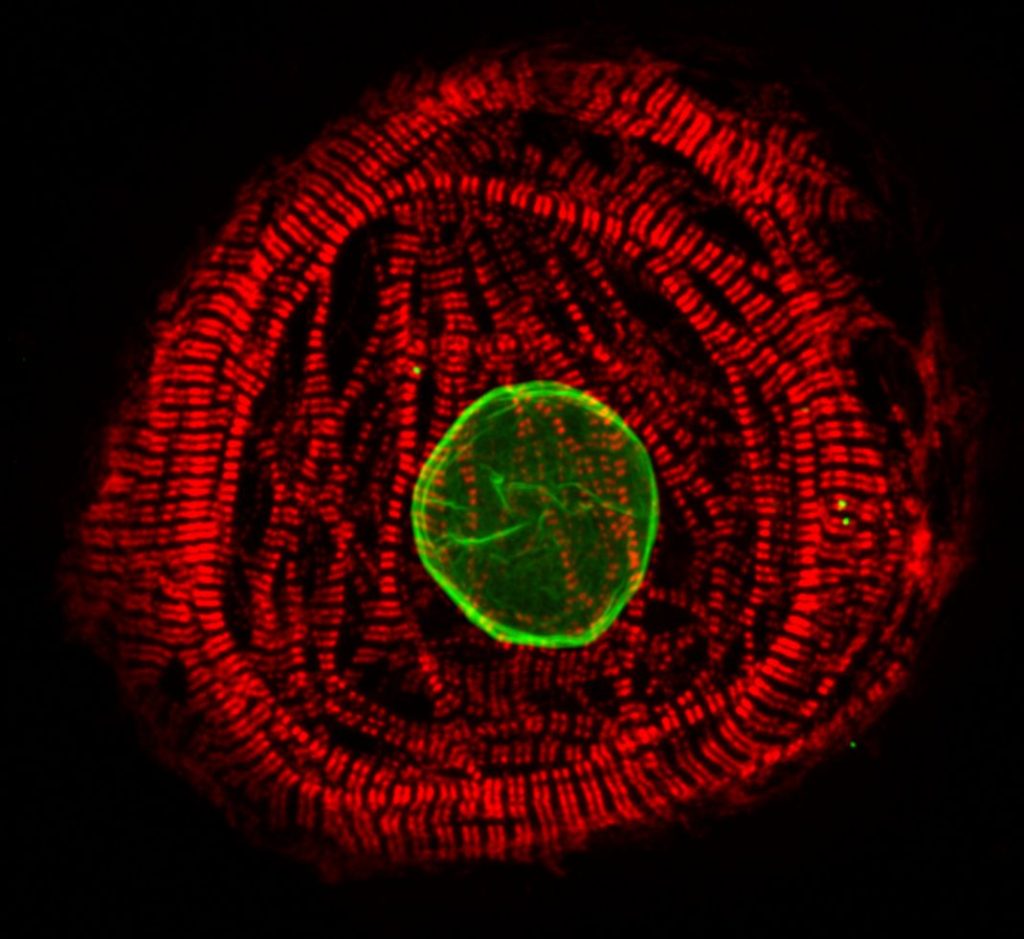
Our research focuses on pathobiology of diseases caused by mutations in the LMNA gene. We are especially interested in understanding how mutations alter lamin assembly, interactions with other nuclear proteins and chromatin, and eventually gene expression profiles and signaling pathways in diseased tissues. In 2015, we identified a previously unknown LMNA mutation (p.L306R) which leads to hyper-assembly of lamin A/C and associates with severe right ventricular cardiomyopathy and premature aging (Alastalo et al., 2015). More recently, we have focused on the founder mutation p.S143P, which accounts for ~7% of all the familial DCM cases in the Finnish population (Kärkkäinen et al., 2004). We have shown that the p.S143P mutation leads to defective filament assembly, nucleoplasmic distribution and aggregation of lamin A/C (West et al., 2016). Elevated ER stress and unfolded protein response (UPR) was detected in the p.S143P mutant patient fibroblasts as well as in p.S143P mutant hiPSC-derived cardiomyocytes (CMs) (West et al., 2016, Shah et al, 2019). These hiPSC-CMs were also found more sensitive to hypoxic stress, showed bradyarrhythmia and an increased number of ventricular type arrhythmias mimicking the clinical features of patients carrying LMNA mutations (Shah et al, 2019).